מה זה 510K ומתי צריך אותו?
הגשת טרום שיווק ל- FDA כדי להוכיח כי המכשיר שישווק הוא שווה ערך באופן מהותי (SE) למכשיר המשווק באופן חוקי שאינו כפוף לאישור PreMarket (PMA).
הגשה חדשה מלאה של 510 (k) נדרשת עבור:
- הצגת מכשיר להפצה מסחרית לראשונה.
- כאשר רוצים להציע שימוש אחר המיועד למכשיר שכבר מופץ מסחרית.
- כאשר בוצעו שינויים במכשיר קיים, אם השינויים עלולים להשפיע באופן משמעותי על בטיחות המכשיר או על יעילותו, או אם המכשיר ישווק לאינדיקציה חדשה או אחרת.
דוגמאות לשינויים שעשויים לדרוש 510 (k):
- שיטת עיקור
- חומר מבני
- שיטת ייצור
- פרמטרים או תנאי שימוש
- חולים או מאפייני בטיחות משתמשים

אינטגרציה של ISO 27001 ו-ISO 42001 — המדריך המעשי לשילוב אבטחת מידע עם ממשל AI בארגון ישראלי. מה ניתן לאחד, מה חייב להישאר נפרד, וכיצד לחסוך 30% ממשאבי ההטמעה. רונית שדה יועצים בע"מ מסבירה.

מ-28 במאי 2026 EUDAMED הוא חובה ליצרני מכשור רפואי בשוק האירופי. צ'קליסט רישום מלא, הדדליין הקריטי הבא ב-28 בנובמבר, ומה קורה אם איחרתם.

הסמכת ISO 42001 כבר מופיעה בדרישות RFP של 40% מהמכרזים לספקי AI באירופה. כך הופכים את תקן ממשל ה-AI מהוצאה רגולטורית למנוף מכירות שמנצח עסקאות.

גז רפואי הוא תרופה לכל דבר. מה דורשים GMP ו-GDP מיצרנים, ממלאים ומפיצים בישראל — ואיך נערכים למבדק בלי הפתעות.

כמה עולה הסמכת ISO 13485 בישראל? מה משפיע על העלות, איך לחסוך, ומתי שווה להביא יועץ. מדריך מעשי ליצרני מכשור רפואי.

QMSR כבר בתוקף, ה-FDA עדכן את הנחיות הסייבר, וה-AI Act מתקרב. מה זה אומר ליצרנים, ליבואנים ולחברות דיגיטל הלת' שכבר עכשיו רוצות להיות מוכנות?

ביום העצמאות 2026 שואלים: האם חברות מדטק ישראליות עצמאיות רגולטורית? QMSR שינה את הכללים — תעודת ISO 13485 כבר לא מספיקה. מדריך מעשי.
תיקון 13 לחוק הגנת הפרטיות, תפוגת ISO 27001:2013, ואיומי סייבר מוגברים — 3 שינויים שמחייבים פעולה מיידית. מדריך מעשי מרונית שדה יועצים בע"מ.
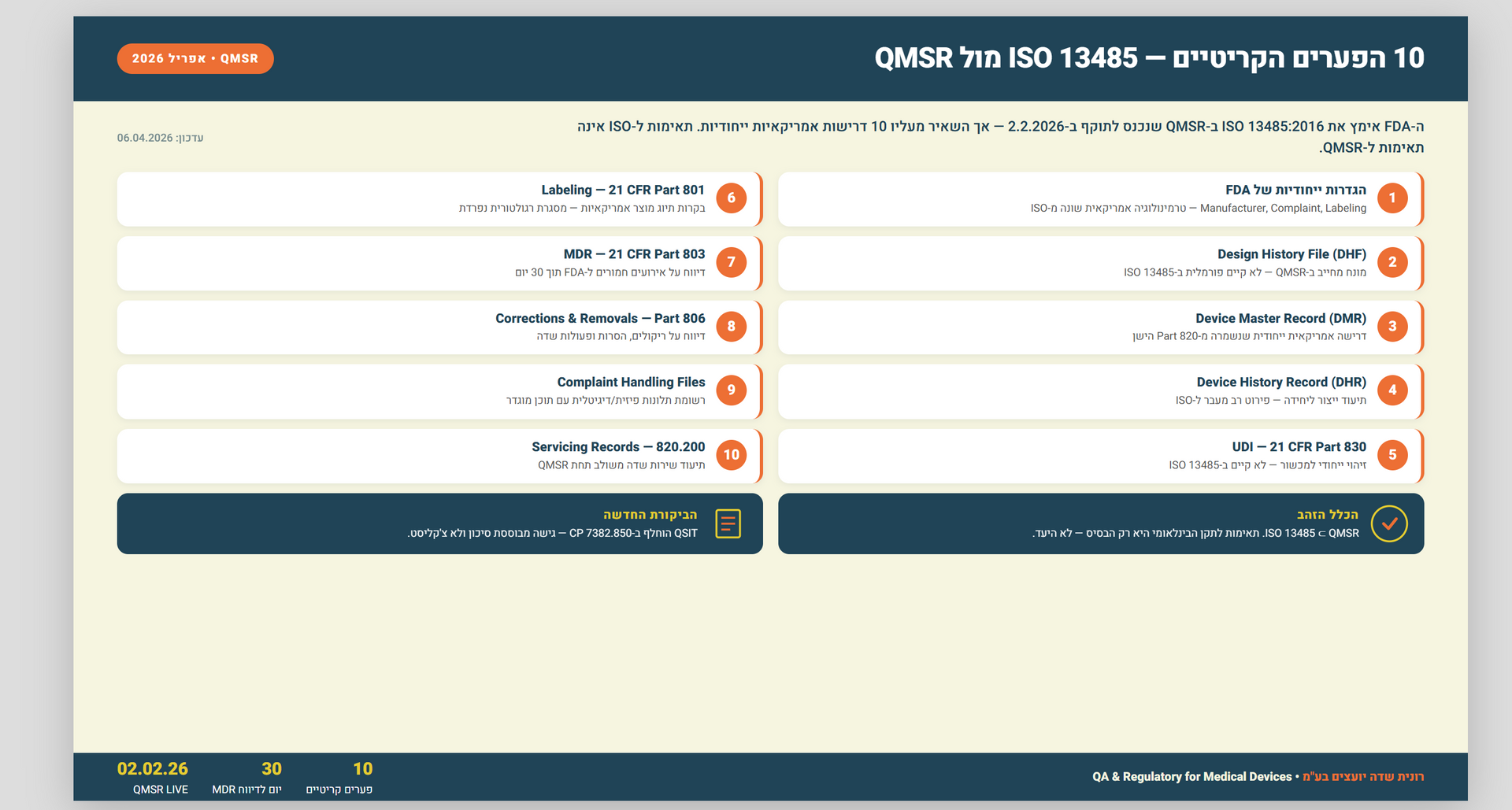
: ISO 13485 מול QMSR — מדריך השוואה מקצועי לכל הפערים בין התקן הבינלאומי לרגולציית ה-FDA החדשה שנכנסה לתוקף בפברואר 2026. עם דוגמאות ופתרונות יישום.
תשובה קצרה: QMSR של ה-FDA, שנכנס לתוקף ב-2 בפברואר 2026, מאמץ את ISO 13485:2016 בהפניה (by reference) אך מוסיף מעליו דרישות אמריקאיות ייחודיות. לכן תאימות מלאה ל-ISO 1348

ניהול איכות תעשייתי מחייב עצירה לחשיבה אחת בשנה. ארבע שאלות מעשיות שמנהל איכות חייב לשאול את עצמו לפני חזרה לעבודה — ולמה פסח הוא הזמן הנכון לעשות זאת.