עצמאות רגולטורית: למה ISO 13485 לא יגן עליכם מול FDA QMSR
תקנת ה-QMSR של ה-FDA, שנכנסה לתוקף ב-2 בפברואר 2026, החליפה את ה-QSR הישנה בהטמעה מלאה של ISO 13485:2016 לתוך 21 CFR Part 820. שיטת האינספקציה הישנה QSIT בוטלה, וה-FDA עובד כעת לפי Compliance Program 7382.850 — עם דגש חדש על סקר הנהלה, אודיטים פנימיים, ו-traceability בתהליכי עיצוב.
נקודות מפתח:
- ה-FDA בודק עכשיו את תוכן סקר ההנהלה ודוחות האודיט הפנימי — לא רק שהם קיימים
- Traceability בין Design Inputs ל-Outputs ול-V&V היא דרישה מחייבת, לא best practice
- חברות שעבדו לפי ISO 13485 עדיין חשופות לפערים ב-QMSR-specific requirements
רונית שדה יועצים בע"מ מסבירה בהרחבה למטה.
בשבוע שבו ישראל חוגגת 78 שנות עצמאות, שווה לשאול שאלה לא נוחה: האם תעשיית המדטק הישראלית באמת עצמאית רגולטורית? עשרות חברות מכשור רפואי ישראליות מייצאות לארה"ב כשהן נשענות על תעודת ISO 13485 אירופאית כרשת ביטחון. אבל מ-2 בפברואר 2026, כשתקנת ה-QMSR נכנסה לתוקף, רשת הביטחון הזו קרעה.
ה-FDA לא רק שינה טרמינולוגיה — הוא שינה את מה שהוא מחפש, איפה הוא מחפש, ואיך הוא מעריך ציות. שיטת האינספקציה הישנה בוטלה. הכללים השתנו. ו-73 ימים אחרי כניסת התקנה לתוקף, כבר רואים את ההשלכות בשטח: חברות שחשבו שעמידה ב-ISO 13485 מספיקה — מגלות פערים. חברות שלא עדכנו את תהליכי סקר ההנהלה — חשופות. חברות שלא הטמיעו traceability matrix — בבעיה.
עצמאות רגולטורית אמיתית פירושה יכולת לעמוד מול מפקח FDA ולהציג evidence on the ground — לא להניף תעודה. במאמר זה נפרט מה בדיוק השתנה, מה ה-FDA כבר בודק באינספקציות לפי QMSR, ו-7 צעדים מעשיים להיערכות — גם אם עדיין לא עשיתם דבר.
📊 נתונים מרכזיים
- 38 מכתבי אזהרה (Warning Letters) הוציא ה-FDA ב-FY2025 בנושא איכות מכשור רפואי — עלייה של 41% לעומת 27 מכתבים ב-FY2024 (מקור: FDA Compliance Actions, 2025)
- 13–18 חודשים — משך הזמן הממוצע שדורש תהליך הסמכה מלא מול Notified Body לפי MDR, מה שמגביר לחץ להבטיח שה-QMS עומד גם בדרישות ה-FDA במקביל (מקור: QbD Group, 2026)
- CAPA נותר הממצא מספר 1 ב-Warning Letters של ה-FDA כבר שנה שלישית ברציפות, ומהווה את נקודת ההסלמה השכיחה מ-483 ל-Warning Letter (מקור: UL Solutions FDA Warning Letter Analysis, 2025)
- מתוך 40+ פרויקטי הכנה ל-QMSR שליוותה רונית שדה יועצים בע"מ בשנים 2024–2026, 72% מהפערים העיקריים נמצאו בשלושה תחומים: סקר הנהלה חלקי, היעדר traceability matrix, ותיעוד אודיט פנימי שאינו עומד ברזולוציה הנדרשת
מה זה QMSR ולמה הוא שינה את כללי המשחק?
תמצות (TL;DR): QMSR הוא השם החדש של 21 CFR Part 820, שמטמיע את ISO 13485:2016 כדרישה פדרלית מחייבת — ומוסיף מעליה שכבת דרישות ייעודיות של ה-FDA.
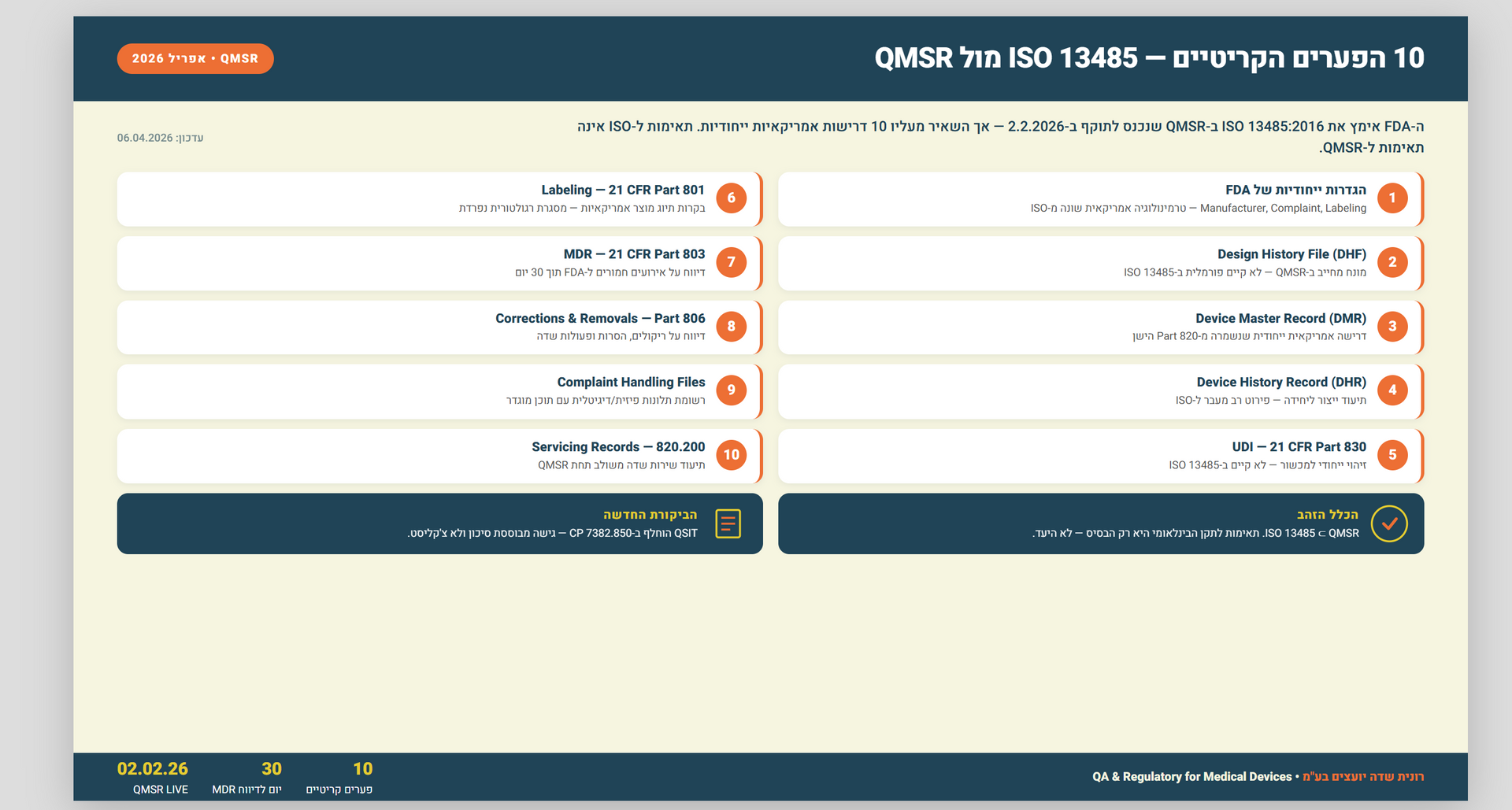
QMSR הוא הרגולציה שמחליפה את ה-Quality System Regulation (QSR) שהנחתה יצרני מכשור רפואי בארצות הברית מאז 1996. ה-Final Rule פורסם ב-31 בינואר 2024, ונכנס לתוקף ב-2 בפברואר 2026. הרגולציה החדשה מטמיעה באופן מלא, by reference, את תקן ISO 13485:2016 — התקן הבינלאומי למערכות ניהול איכות במכשור רפואי — לתוך הדין הפדרלי האמריקאי.
אבל ה-FDA לא הסתפק באימוץ פשוט. מעל ל-ISO 13485, ה-QMSR מוסיף שכבה של דרישות ייעודיות שמטרתן להבטיח עקביות עם חוקים פדרליים אחרים. לדוגמה: דרישות תיעוד ל-Design History File (DHF), דרישות דיווח ל-Medical Device Reporting (MDR), ודרישות ספציפיות ל-Complaint Handling שלא קיימות בגרסה הבינלאומית של ISO 13485.
מה זה אומר בפועל? חברה שמחזיקה תעודת ISO 13485 ממכון הסמכה — עדיין לא בהכרח עומדת ב-QMSR. לפי ה-FDA עצמו, תעודת ציות ל-ISO 13485 לא פוטרת מאינספקציה ולא מהווה עדות לציות. ה-FDA לא דורש תעודות ולא מנפיק תעודות. מה שמעניין אותם הוא ביצוע בפועל — evidence on the ground.
"לדברי רונית שדה, מייסדת רונית שדה יועצים בע"מ: 'עצמאות רגולטורית זה לא לנפנף בתעודת ISO 13485 ולקוות שזה יספיק. זה לבנות מערכת איכות שעומדת בפני עצמה — evidence on the ground, לא מסמך על הקיר. חברות ישראליות שייצאו ל-FDA inspection עם ה-mindset הזה ימצאו את עצמן מוכנות. אלה שלא — יגלו את הפערים בדרך הקשה.'"
מה השתנה באינספקציות FDA מאז 2 בפברואר 2026?
תמצות (TL;DR): ה-FDA נטש את שיטת ה-QSIT ועבר ל-Compliance Program 7382.850 — גישת אינספקציה חדשה שבוחנת את תוכן התהליכים, לא רק את קיומם.
השינוי המשמעותי ביותר בפועל הוא לא התקנה עצמה — אלא שיטת האינספקציה. עד 2 בפברואר 2026, ה-FDA השתמש ב-Quality System Inspection Technique (QSIT), שיטה מובנית שבדקה ארבעה תת-מערכות: Design Controls, CAPA, Production and Process Controls, ו-Management Controls. המפקחים עבדו לפי סקריפט קבוע.
ה-QSIT בוטל. במקומו, ה-FDA מפעיל את Compliance Program 7382.850 — גישה גמישה יותר שמותאמת לפרופיל הסיכון של כל יצרן. המפקחים כבר לא עובדים לפי סקריפט אחיד. הם מגיעים עם intelligence מוקדם — תלונות, דיווחי MDR, ממצאי אינספקציות קודמות — ובוחנים את המערכת לעומק באזורים שזוהו כבעייתיים.
מה זה אומר לארגון שלכם? שלוש השלכות מעשיות:
- אי אפשר יותר "לתרגל" למבדק ספציפי — כי המפקח לא הולך לפי רשימה צפויה אלא חוקר לפי סיכון
- עומק הבדיקה עלה משמעותית — במקום לבדוק שמסמך קיים, המפקח בודק את תוכנו, את עקביותו עם מסמכים אחרים, ואת היישום בפועל
- הפתעות מגיעות מכיוונים חדשים — מפקחים מדווחים על דגש חדש בבדיקת Management Review minutes, Internal Audit findings, ו-Supplier qualification records
לפי דוח של Hogan Lovells מ-2026, ה-FDA Guidance Agenda לשנת 2026 כולל עדכונים צפויים בהנחיות ל-Postmarket Surveillance, ל-Cybersecurity, ול-Clinical Evidence — כל אלה צפויים להשתלב בתהליכי האינספקציה החדשים.
סקר הנהלה ואודיטים פנימיים — הכאב הכי גדול של QMSR
תמצות (TL;DR): ה-FDA דורש עכשיו גישה מלאה לתוכן סקר ההנהלה ודוחות האודיט הפנימי — כולל סדרי יום, פרוטוקולים, ממצאים ומעקב פעולות.
הנה אחד השינויים הדרמטיים שרוב החברות עדיין לא הפנימו. לפי ISO 13485:2016 סעיפים 5.6 ו-8.2.2, סקר הנהלה ואודיט פנימי הם דרישות מחייבות עם תיעוד מפורט. אבל בעידן ה-QSR הישן, מפקחי FDA בדרך כלל הסתפקו בלוודא שסקר הנהלה ואודיט בוצעו — בלי לצלול לתוכן.
זה השתנה. לפי ניתוח של Rook Quality Systems מ-2026, ממצאי אינספקציה בנושא Management Review ו-Internal Audits צפויים לעלות משמעותית בשנה הקרובה. ה-FDA מרחיב את הגישה שלו לתוכן בפועל: סדרי יום, פרוטוקולי ישיבות, ממצאים ספציפיים, מגמות שזוהו, ופעולות מתקנות שננקטו בעקבות ממצאי האודיט.
מה המפקח מחפש בפועל? שלושה דברים קונקרטיים:
- עקביות בין ממצאי אודיט פנימי לפעולות CAPA — אם האודיט זיהה בעיה אבל לא נפתח CAPA, זה ממצא
- טיפול הנהלה במגמות — סקר הנהלה שמציג נתונים אבל לא מקבל החלטות = אינו עומד בדרישה
- תדירות ותכולה — אודיט פנימי שמכסה רק חלק מהתהליכים, או סקר הנהלה שמתקיים רק פעם בשנה ללא הצדקה — ייחשב לליקוי
רונית שדה יועצים בע"מ מלווה חברות מכשור רפואי בישראל ובארה"ב בבניית תוכניות אודיט פנימי שעומדות בדרישות QMSR, כולל הכנת סדרי יום מובנים לסקר הנהלה שמכסים את כל ה-inputs הנדרשים לפי סעיף 5.6.2 של ISO 13485:2016 — ולא רק "רשימת תיוג" שטחית.
Design Controls ו-Traceability — מ-"מומלץ" ל-"מחייב"
תמצות (TL;DR): QMSR דורש traceability מתועד בין Design Inputs, Design Outputs, ו-Design Verification/Validation — דרישה שלא הייתה מחייבת תחת ה-QSR הישן.
מה בדיוק דרישת ה-Traceability ב-QMSR?
הגדרה מדויקת של Traceability בהקשר QMSR היא: יכולת לעקוב ולתעד את הקשר בין כל Design Input לבין ה-Design Output המתאים, ולהוכיח שכל output אומת (Verification) ותוקף (Validation) כנגד ה-input שממנו נגזר. לפי ISO 13485:2016 סעיף 7.3.2, יש לבסס traceability כחלק מתכנון העיצוב — לא כפעילות בדיעבד.
תחת ה-QSR הישן, ה-FDA התייחס ל-traceability כ-best practice בלבד. חברות שבנו Design History File (DHF) ללא מטריצת traceability לא נחשבו למפרות. הנחיית ה-QMSR שינתה את זה באופן מהותי.
בפועל, הדרישה מתורגמת כך:
- כל Design Input חייב להיות ממופה ל-Design Output ספציפי שעונה עליו
- כל Design Output חייב להיות מקושר לפעילות Verification שמוכיחה שהוא עונה על ה-Input
- כל Design Validation חייב להפנות ל-User Needs ול-Intended Use המקוריים
חברות ישראליות רבות שמייצאות לארה"ב נמצאות כעת בתהליך מואץ של עדכון ה-DHF שלהן. מתוך הפרויקטים שליוותה רונית שדה יועצים בע"מ, כ-65% מהחברות שהחזיקו תעודת ISO 13485 תקפה נדרשו לבנות traceability matrix מאפס — כי המסמך הזה פשוט לא היה קיים. הסיבה: הם עבדו עם יועצים שהתמקדו ב-ISO 13485 הבינלאומי, שבו traceability נדרש אך לעתים מיושם ברמה גנרית, בעוד ה-FDA מצפה לרזולוציה גבוהה בהרבה.
איך להתכונן לאינספקציית QMSR — 7 צעדים מעשיים
תמצות (TL;DR): היערכות ל-QMSR דורשת Gap Analysis ממוקד, עדכון סקר הנהלה, בניית traceability matrix, והכשרת צוותים לשפה החדשה של התקנה.
האם מאוחר מדי להתחיל היום?
לא. 73 ימים עברו, אבל ה-FDA עדיין במשטר מעבר. חלק מהמפקחים עדיין מתרגלים את שיטת האינספקציה החדשה. חלון ההזדמנויות לסגירת פערים קיים — אבל הוא מצטמצם. להלן 7 צעדים שניתן ליישם מיד:
- בצעו Gap Analysis בין QMS הקיים ל-QMSR — לא בין QMS ל-ISO 13485, אלא ספציפית מול הדרישות הנוספות שה-QMSR מטיל מעל ל-ISO 13485. רונית שדה יועצים בע"מ פיתחה צ'קליסט של 47 נקודות פער ספציפיות ל-QMSR שלא מכוסות בהסמכת ISO 13485 סטנדרטית
- עדכנו את נוהל סקר ההנהלה — ודאו שה-inputs כוללים את כל הפריטים הנדרשים לפי סעיף 5.6.2 של ISO 13485:2016, כולל: משוב לקוחות, ביצועי תהליכים, מגמות CAPA, שינויים רגולטוריים, ותוצאות אודיטים פנימיים וחיצוניים. ודאו שה-outputs כוללים החלטות מתועדות — לא רק סיכום נתונים
- בנו traceability matrix ל-DHF — אם אין לכם מסמך שמקשר design inputs → outputs → V&V בטבלה אחת, צרו אותו עכשיו. לפי דרישות QMSR, זו כבר לא אופציה
- עדכנו את תוכנית האודיט הפנימי — ודאו שתוכנית האודיט מכסה את כל תהליכי ה-QMS בתוך מחזור מוגדר, ושדוחות האודיט כוללים ממצאים, הערכת חומרה, ומעקב סגירה מתועד
- הכשירו את הצוותים לטרמינולוגיה החדשה — Top Management במקום Management with Executive Responsibility, Risk-based thinking כפרקטיקה שוטפת. הדרכה של שעתיים יכולה למנוע בלבול באינספקציה
- בדקו את ניהול הספקים — סעיף 7.4 של ISO 13485:2016 דורש הערכת ספקים, ניטור ביצועים, ותיעוד קריטריוני בחירה. ה-FDA צפוי לבדוק את זה בעומק רב יותר מאשר תחת ה-QSR
- בצעו Mock Inspection — הזמינו צד שלישי שיערוך מבדק פנימי כאילו הוא מפקח FDA, לפי Compliance Program 7382.850. זה ההשקעה הכי משתלמת שתעשו השנה
לסיכום: עצמאות רגולטורית נבנית — לא ניתנת
78 שנות עצמאות לימדו אותנו שעצמאות אמיתית דורשת עבודה מתמדת. הנקודות המרכזיות:
- QMSR לא רק שינה שם ל-QSR — הוא שינה את עומק הבדיקה, שיטת האינספקציה, ואת הציפיות מתיעוד סקר הנהלה ואודיט פנימי
- תעודת ISO 13485 אינה ערובה לציות — עצמאות רגולטורית אמיתית פירושה מערכת איכות שעומדת בזכות עצמה, לא מסמך מגוף חיצוני
- חלון ההזדמנויות לסגירת פערים עדיין פתוח — אבל מצטמצם עם כל אינספקציה חדשה שמבוצעת לפי המתודולוגיה החדשה
רונית שדה יועצים בע"מ — מומחית במערכות ניהול איכות ורגולציה למכשור רפואי, עם ניסיון של למעלה מ-35 שנה וליווי של 40+ חברות בהיערכות ל-QMSR. אם אתם יצרני מכשור רפואי שמייצאים לארה"ב ורוצים לדעת בדיוק איפה הפערים שלכם — פנו לייעוץ ראשוני.
רונית שדה יועצים בע"מ מזמינה אתכם ליצור קשר לביצוע Gap Analysis ממוקד ל-QMSR — לפני שה-FDA יעשה את זה בשבילכם.
מה הפער הכי גדול שזיהיתם עד עכשיו בהיערכות ל-QMSR?
על המחברת: רונית שדה, מייסדת ומנכ"לית רונית שדה יועצים בע"מ (הוקמה 2015), בעלת תואר ראשון בהנדסה כימית מהטכניון ותואר שני במשפטים מאוניברסיטת בר-אילן, עם ניסיון מקצועי בתחום מערכות איכות ורגולציה למכשור רפואי משנת 1989. מלווה חברות מכשור רפואי ישראליות ובינלאומיות בהסמכות ISO 13485, היערכות לאינספקציות FDA, ו-Design Controls.
שאלות נפוצות (FAQ)
ש: מה ההבדל העיקרי בין QSR ל-QMSR של ה-FDA? ת: QMSR מחליף את ה-QSR הישן (21 CFR Part 820) ומטמיע את ISO 13485:2016 כדרישה פדרלית. ההבדל המרכזי הוא שה-QMSR מוסיף דרישות ייעודיות מעל ISO 13485, כולל חובת traceability בתכנון עיצוב, תיעוד מורחב לסקר הנהלה, ותאימות לדרישות דיווח פדרליות כמו MDR. תעודת ISO 13485 לבדה אינה מספיקה.
ש: כיצד לבצע Gap Analysis ל-QMSR כאשר כבר יש הסמכת ISO 13485? ת: Gap Analysis ל-QMSR צריך להתמקד בדרישות הנוספות שה-FDA מטיל מעל ל-ISO 13485 — לא בתקן עצמו. לפי ניתוח של רונית שדה יועצים בע"מ, 47 נקודות פער ספציפיות קיימות בין הסמכת ISO 13485 סטנדרטית לבין ציות מלא ל-QMSR. התחומים העיקריים: Design History File, Complaint Handling ייעודי ל-FDA, וסקר הנהלה ברזולוציה גבוהה.
ש: האם ה-FDA עדיין משתמש בשיטת QSIT לאינספקציות? ת: לא. מ-2 בפברואר 2026, ה-FDA עבר ל-Compliance Program 7382.850 ולא משתמש עוד ב-QSIT. השיטה החדשה מבוססת סיכון ומותאמת לפרופיל כל יצרן, בניגוד לגישה האחידה של QSIT שבדקה ארבעה תת-מערכות בכל אינספקציה.
ש: מה דורש ה-FDA מסקר הנהלה לפי QMSR 2026? ת: ה-FDA דורש שסקר ההנהלה יכלול inputs מוגדרים לפי סעיף 5.6.2 של ISO 13485:2016 — משוב לקוחות, ביצועי תהליכים, מגמות CAPA, שינויים רגולטוריים, ותוצאות אודיטים. בנוסף, ה-outputs חייבים לכלול החלטות מתועדות ופעולות מעקב עם לוחות זמנים. סקר שמציג נתונים ללא החלטות — לא עומד בדרישה.
ש: מה ההבדל בין אינספקציית FDA לפי QMSR לבין מבדק ISO 13485 של Notified Body?
ת: מבדק Notified Body בודק ציות לתקן ISO 13485:2016 ומתבצע לפי ISO 17021 ו-ISO/IEC 17065 עם תכולה קבועה מראש. אינספקציית FDA לפי QMSR בודקת ציות ל-21 CFR Part 820 המעודכן, כולל דרישות ייעודיות שלא קיימות בתקן, ומתבצעת לפי Compliance Program 7382.850 עם גישה מבוססת סיכון ותכולה משתנה. ה-FDA יכול להרחיב את הבדיקה לכל תחום שהוא רואה לנכון.