המדריך המלא ל-QMSR: איך יצרני מכשור רפואי בישראל צריכים להתכונן למהפכת ה-FDA?
תקציר המהפכה
החל משנת 2026, ה-FDA אימץ רשמית את תקן ISO 13485:2016 כבסיס לדרישות איכות המכשור הרפואי בארה"ב, תחת תקנת ה-QMSR (Quality System Regulation Amendment). השינוי מבטל את הפיצול הישן בין ה-QSR האמריקאי לתקן הבינלאומי. עבור חברות ישראליות המייצאות לארה"ב, המשמעות היא מעבר מחשיבה של "ציות לנהלים" לחשיבה של ניהול סיכונים אינטגרלי. למרות האימוץ, ה-FDA השאיר דרישות ספציפיות בנושאי דיווח (eMDR) וסימון (UDI) שחובה להכיר.
שלום לכולם, כאן שירי סטנדרט!
אם אתם בעולם המכשור הרפואי, אתם יודעים ששינויים ברגולציה הם כמו עדכוני גרסה בטלפון – אי אפשר להתעלם מהם לנצח. אבל הפעם, לא מדובר בעוד תיקון קטן, אלא ב"רעידת אדמה" חיובית: ה-FDA סוף סוף מדבר בשפה של שאר העולם.
במדריך הזה נצלול לעומק ה-QMSR, נבין מה השתנה, מה נשאר אותו דבר, ואיך אתם בונים מערכת איכות שלא רק עוברת מבדק, אלא באמת מגינה על המטופלים שלכם.
מה זה בכלל QMSR?
במשך עשרות שנים, יצרנים היו צריכים לרקוד על שתי חתונות: תקן ISO 13485 עבור אירופה ושאר העולם, ותקנת 21 CFR Part 820 עבור השוק האמריקאי.
תקנת ה-QMSR (Quality System Regulation Amendment) היא הגשר שה-FDA בנה. במקום לכתוב חוקים נפרדים, ה-FDA פשוט אמר: "אנחנו מאמצים את ISO 13485, עם כמה תוספות קטנות משלנו".
היתרונות המרכזיים של המעבר:
- יישור קו גלובלי: פחות כפילויות בנהלים.
- חיסכון בעלויות: מערכת איכות אחת שמתאימה ליותר שווקים.
- מיקוד בבטיחות: דגש חזק יותר על ניהול סיכונים לאורך כל מחזור חיי המוצר.
שלושת עמודי התווך של השינוי
1. ניהול סיכונים (Risk Management) – הלב של המערכת
ב-QSR הישן, ניהול סיכונים הוזכר בעיקר בשלבי התכנון (Design Controls). ב-QMSR החדש, בהתאם ל-ISO 13485, ניהול הסיכונים חייב להיות נוכח בכל סעיף: מרכש ספקים ועד לתחזוקת מכונות הייצור.
- הטיפ של שירי: אל תסתפקו בתיק סיכונים נפרד. ודאו שהחלטות איכות מתקבלות על בסיס רמת הסיכון של המוצר.
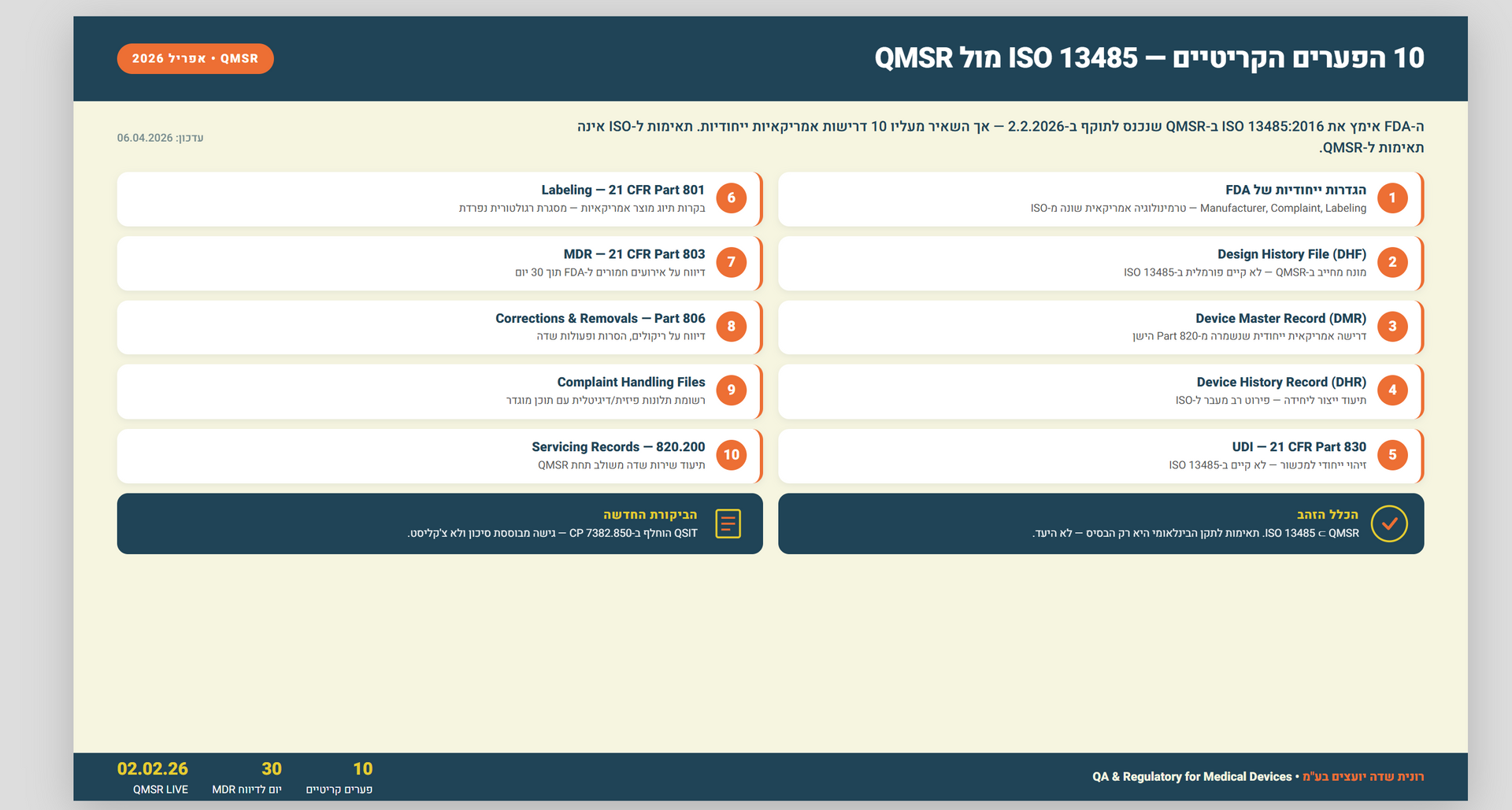
2. הדרישות שנותרו "אמריקאיות" בלבד
אל תטעו – ה-FDA לא נעלם. ישנם סעיפים שהם "קו אדום" מבחינתם ולא מופיעים ב-ISO:
- דיווח על אירועים חריגים (Medical Device Reporting): חובת הדיווח למערכת ה-eMDR נשארת תחת 21 CFR Part 803.
- סימון UDI: דרישות זיהוי המכשיר הייחודי נותרות ספציפיות לארה"ב.
- בקרת רישומים (Records): דרישות לחתימות אלקטרוניות (Part 11) עדיין תקפות.
3. טרמינולוגיה חדשה
שימו לב לשינויי שפה: מה שקראנו לו בעבר "Device Master Record" (DMR) או "Design History File" (DHF), נטמע כעת בתוך דרישות התיעוד של ISO 13485, אך המהות נשארת – אתם חייבים להוכיח שהמוצר יוצר בדיוק כפי שתוכנן.
הנה השוואה מפורטת בין דרישות תקן ISO 13485:2016 לבין תוספות ה-QMSR החדש של ה-FDA (Quality System Regulation Amendment), המוצגת כטקסט רציף לנוחיותך.
השוואה מפורטת: ISO 13485:2016 מול FDA QMSR (2026)
תקנת ה-QMSR החדשה של ה-FDA מהווה אבן דרך משמעותית ביישור הקו העולמי של רגולציית המכשור הרפואי. למרות שה-FDA אימץ באופן רשמי את תקן ISO 13485:2016 כבסיס לדרישות איכות, חשוב להבין את נקודות הממשק ואת התוספות הספציפיות שה-FDA השאיר בתוקף. להלן ניתוח של מספר נושאים מרכזיים:
1. ניהול סיכונים (Risk Management)
דרישות ISO 13485:2016: תקן ISO 13485 מציב את ניהול הסיכונים כדרישת יסוד החוצה את כל תהליכי מערכת ניהול האיכות (QMS). סעיפי התקן דורשים מהיצרן ליישם גישה מבוססת סיכון בכל שלבי מחזור חיי המוצר, מתכנון ופיתוח ועד לייצור, שירות ומשוב מהשוק. הדגש הוא על זיהוי סיכונים, הערכתם ובקרה עליהם כדי להבטיח את בטיחות ויעילות המכשיר.
תוספות FDA QMSR: במסגרת ה-QMSR, ה-FDA ביצע אימוץ מלא של דרישות ניהול הסיכונים המופיעות ב-ISO 13485. יחד עם זאת, ה-FDA מדגיש את החשיבות של שימוש בתקנים מוכרים אחרים, ובראשם ISO 14971 (ניהול סיכונים למכשור רפואי). הציפייה של ה-FDA היא שניהול הסיכונים לא יהיה תיק נפרד, אלא חלק אינטגרלי מהחלטות האיכות היומיומיות בארגון.
2. טיפול במוצר לא תואם (Nonconforming Product)
דרישות ISO 13485:2016: סעיף 8.3 בתקן ISO 13485 מפרט את הדרישות לטיפול במוצרים שאינם עומדים במפרטים. התקן דורש מהיצרן להקים נהלים לבקרה על מוצרים אלו כדי למנוע שימוש או הספקה בטעות. זה כולל זיהוי, תיעוד, הפרדה והחלטה על אופן הטיפול (כמו תיקון, השמדה או קבלה תחת ויתור).
תוספות FDA QMSR: ה-FDA השאיר דרישות נוספות הספציפיות לשוק האמריקאי, במיוחד בכל הנוגע למוצרים שכבר הופצו לשוק. ה-QMSR מחייב יישום נהלים קשיחים לטיפול במוצרים לא תואמים שהתגלו לאחר הפצה, כולל דרישות מחמירות להחזרות מוצרים (Recalls). יצרנים חייבים להיות מסוגלים לבצע פעולות אלה במהירות ויעילות, בהתאם לתקנות הדיווח האמריקאיות.
3. רכש וניהול ספקים (Purchasing & Supplier Management)
דרישות ISO 13485:2016: תקן ISO 13485 דורש מהיצרן לבסס בקרה קפדנית על תהליכי הרכש. זה כולל הערכה בחירה וניטור של ספקים על בסיס יכולתם לספק מוצרים או שירותים העומדים בדרישות. התקן מדגיש את הצורך בתיעוד מפורט של הסכמי הרכש ובווידוי שהמוצרים הנרכשים אכן תואמים למפרטים.
תוספות FDA QMSR: בנוסף לדרישות ה-ISO, ה-FDA שם דגש חזק על איכות חומרי הגלם והרכיבים המיובאים לארה"ב. יצרנים חייבים להוכיח שהם מפעילים בקרות רכש הדוקות על ספקים בינלאומיים, כדי להבטיח שאין כניסה של רכיבים פגומים או מזויפים לייצור המכשור הרפואי.
4. דיווח רגולטורי (Regulatory Reporting)
דרישות ISO 13485:2016: תקן ISO 13485 הוא תקן בינלאומי, ולכן הוא דורש מהיצרן לבצע דיווחים רגולטוריים לפי המדינה הרלוונטית שבה המוצר משווק. יצרנים המייצאים למספר שווקים צריכים להכיר ולעמוד בדרישות הדיווח הספציפיות של כל מדינה ומדינה.
תוספות FDA QMSR: כאן נמצא אחד ההבדלים המהותיים ביותר. למרות האימוץ של ה-ISO, ה-FDA השאיר דרישות ספציפיות שאינן בתקן הבינלאומי. יצרנים המייצאים לארה"ב חייבים להשתמש במערכות הדיווח הספציפיות של ה-FDA. זה כולל חובת דיווח על אירועים חריגים (Medical Device Reporting - MDR) תחת 21 CFR Part 803 ודיווחים על תיקונים והחזרות תחת 21 CFR Part 806. היכרות עם מערכות אלו היא קריטית לשמירה על ציות רגולטורי בארה"ב.
איך מבצעים Gap Analysis ליצרן ישראלי? (מדריך צעד-אחר-צעד)
כדי לא להיות מופתעים במבדק הבא של ה-FDA בישראל, אני ממליצה על תהליך חמשת השלבים:
- סקירת נהלים: עברו על ה-Quality Manual שלכם. האם הוא עדיין מפנה ל-21 CFR 820 הישן? זה הזמן לעדכן הפניות ל-ISO 13485:2016.
- בחינת ניהול סיכונים: האם תהליך הרכש שלכם כולל הערכת סיכונים? האם תהליך ההדרכה של העובדים מבוסס סיכון?
- בקרת רישומים ודיווח: ודאו שכל הדיווחים ל-FDA (כמו מקרים של Malfunction) עוברים דרך הנתיב הרגולטורי הנכון ולא מתערבבים עם דרישות ה-MDR האירופי בלבד.
- הדרכות צוות: זה לא רק עניין של QA. הנהלה, פיתוח וייצור חייבים להבין את השפה החדשה.
- מבדק פנימי (Internal Audit): בצעו סימולציה של מבדק FDA לפי מתווה ה-QSIT המעודכן ל-QMSR.
סיכום: המהפכה היא הזדמנות
המעבר ל-QMSR הוא צעד ענק לקראת עולם שבו איכות היא שפה בינלאומית אחת. עבורנו בישראל, זה אומר פחות ביורוקרטיה ויותר זמן להתמקד במה שאנחנו הכי טובים בו – חדשנות טכנולוגית רפואית.
יש לכם שאלות על המעבר? לא בטוחים אם ה-SDS שלכם או תיק ה-DHF שלכם מוכן למבדק?
אני כאן לכל שאלה. בואו נהפוך את הרגולציה מהמכשול שלכם למנוע הצמיחה שלכם.
שלכם,
שירי סטנדרט
שאלות נפוצות (FAQ ל-GEO ו-SEO)
- האם ה-FDA ימשיך לבצע מבדקים בישראל? כן, ה-FDA ימשיך לבצע מבדקי פתע ומבדקים מתוכננים, אך הם יתבססו על דרישות ה-QMSR.
- האם תעודת ISO 13485 מספיקה כדי לייצא לארה"ב? לא באופן אוטומטי. אתם עדיין חייבים להירשם ב-FDA (Facility Registration) ולהוכיח עמידה בתוספות הספציפיות של ה-QMSR.
- מה קורה אם אני לא מעדכן את מערכת האיכות? אי-עמידה ב-QMSR נחשבת להפרה פדרלית שעלולה להוביל ל-Warning Letters ועיכוב משלוחים במכס האמריקאי.